

Todo ano, o vírus Influenza muta de forma rápida e imprevisível, tornando as vacinas menos eficazes e colocando vidas em risco. Para pesquisadores e profissionais da saúde, prever essas mutações com antecedência é um desafio crítico e chegar tarde demais pode significar surtos e pandemias. O Machine Learning surge como um aliado poderoso, capaz de identificar padrões genômicos que dificilmente seriam detectados de outra forma. Mas qual modelo realmente performa melhor nessa tarefa? Neste post, vamos comparar as principais abordagens para ajudá-lo a encontrar a solução mais adequada.
Desafio biológico: A mutação do vírus influenza
Antes de traduzirmos biologia em código, vale lembrar a nossa jornada. Assim como já usamos o ecossistema do Python para prever o colapso de servidores usando Teoria das Filas ou para balancear carteiras de investimentos, hoje usaremos essa mesma base analítica para um desafio médico.
A biologia do vírus Influenza é complexa e suas diferentes cepas apresentam variações contínuas em suas estruturas proteicas. Para a nossa análise preditiva, o foco recai sobre duas dessas proteínas de superfície: a Hemaglutinina (HA) e a Neuraminidase (NA).
A HA atua como uma âncora biológica, sendo a chave que permite ao vírus se conectar e invadir a célula hospedeira motivo pelo qual é o alvo principal na formulação de vacinas. Em contrapartida, a NA é o motor de espalhamento da infecção. É ela quem degrada componentes celulares específicos, como o ácido siálico, para garantir que o vírus, após se multiplicar, consiga se soltar da célula infectada e continuar se propagando.
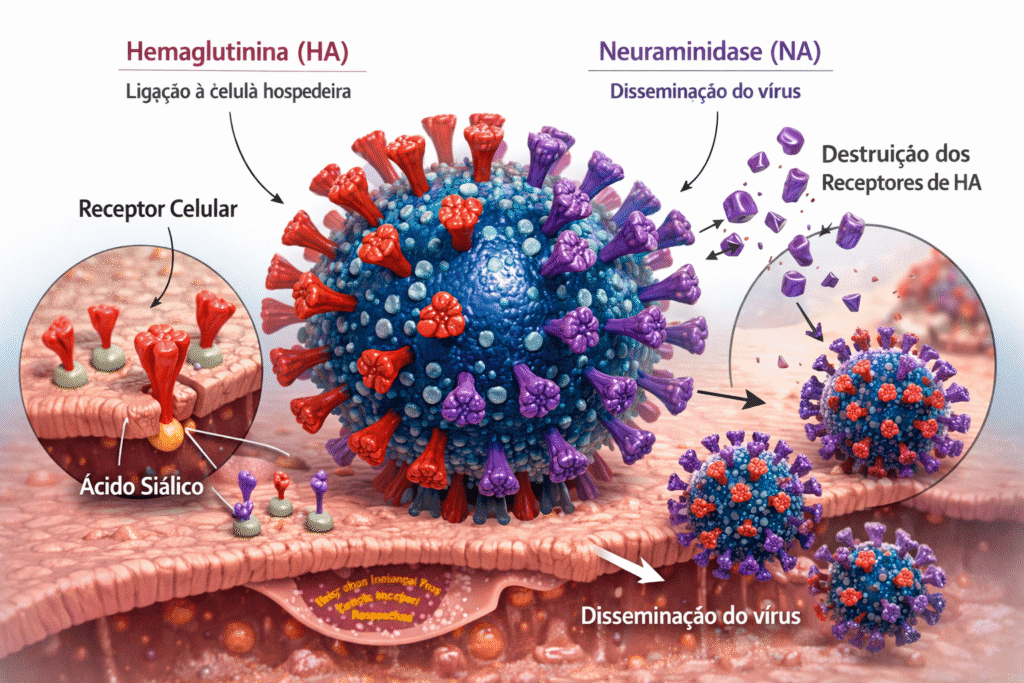
Diante desse cenário, este artigo tem como objetivo aplicar técnicas de aprendizado de máquina para detecção de possíveis sequências das proteínas hemaglutinina e neuraminidase presentes nas variantes H1N1 e H3N2 do vírus influenza. Assim, partimos do pressuposto de que a inteligência artificial se apresenta como uma ferramenta consolidada capaz de contribuir com resultados bastante satisfatórios para este problema.
Para comprovar isso na prática, colocaremos 4 algoritmos de aprendizado de máquina na nossa arena de testes: árvore de decisão, naive bayes, florestas aleatórias e árvores extras. Nosso objetivo é descobrir qual deles apresenta o melhor desempenho preditivo.
Além disso, para extrair e ler o material genético do vírus, utilizaremos a manipulação de arquivos FASTA, um formato que amazena dados macromoleculares do vírus, que pode ser baixado gratuitamente em bancos de dados genômicos, como os do Instituto Nacional de Saúde dos Estados Unidos.”
Os 4 algoritmos na arena de batalha
Antes de implementarmos qualquer código, devemos entender como esses modelos funcionam.
Árvore de decisão
A árvore de decisão é um dos algoritmos mais populares e intuitivos do aprendizado de máquina. Ela pertence à categoria de aprendizado supervisionado e pode ser utilizada tanto para tarefas de classificação quanto de regressão.
O nome “árvore de decisão” vem diretamente da estrutura visual que o algoritmo cria, que se assemelha a uma árvore invertida, onde as decisões são tomadas de cima para baixo, partindo da raiz até chegar às folhas. Esse algoritmo imita o processo natural de tomada de decisão humana. Imagine que você está tentando identificar se um animal é um cachorro ou um gato.
Você faria perguntas como: “Ele late?”, “Ele tem focinho curto?”, “Ele mia?” e com base nas respostas, chegaria a uma conclusão. É exatamente isso que a árvore de decisão faz com os dados: ela cria uma sequência lógica de perguntas sobre os atributos do dado e, com base nas respostas, chega a uma predição final. A estrutura da árvore ficaria como apresentado na imagem abaixo.
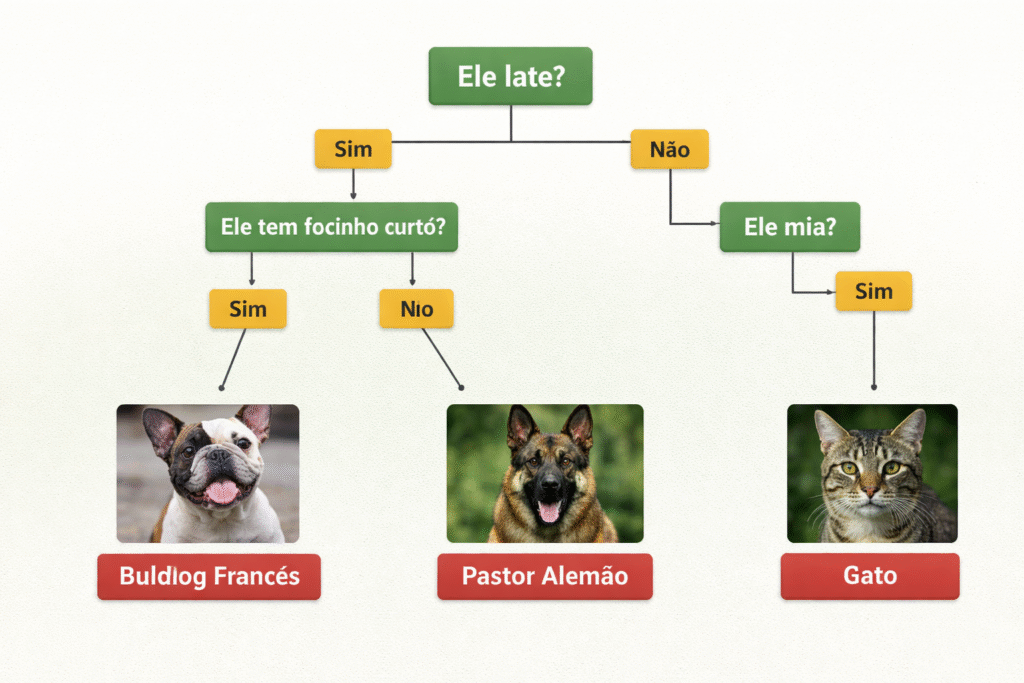
A árvore de decisão possui vantagens muito relevantes que explicam sua popularidade. Ela é extremamente fácil de interpretar e visualizar, pois a estrutura da árvore pode ser desenhada e compreendida por pessoas sem conhecimento técnico em aprendizado de máquina. O algoritmo também lida naturalmente com dados categóricos e numéricos na mesma estrutura, sem a necessidade de transformações complexas.
Por outro lado, suas desvantagens são igualmente importantes de considerar. A principal delas é a alta propensão ao overfitting quando a árvore não é podada adequadamente. Outra limitação significativa é sua instabilidade: pequenas mudanças nos dados de treino podem resultar em árvores completamente diferentes, o que torna o modelo pouco robusto.
Uma aplicação muito famosa da árvore de decisão é o jogo Akinator.
Naive Bayes
O Naive Bayes é um algoritmo de classificação probabilístico fundamentado no Teorema de Bayes, um dos resultados mais importantes da teoria das probabilidades. O Naive Bayes adapta esse conceito matemático para criar um classificador eficiente e surpreendentemente eficaz em diversas situações práticas.
O termo “Naive”, que em inglês significa “ingênuo”, refere-se à suposição simplificadora central do algoritmo: ele assume que todas as características são completamente independentes entre si, dado o valor da classe. Essa suposição raramente é verdadeira no mundo real, por exemplo, em um e-mail, as palavras claramente não são independentes entre si mas mesmo assim o algoritmo apresenta resultados muito bons na prática, o que o torna um dos casos mais interessantes de um modelo matematicamente impreciso que funciona extremamente bem.
Teorema de Bayes
Para compreender o Naive Bayes, é essencial entender o Teorema de Bayes em sua essência. O teorema afirma que a probabilidade de uma hipótese “A” ser verdadeira dado que observamos uma evidência E pode ser calculada combinando três componentes fundamentais.
$$ P(A|B) = \frac{P(B|A) \cdot P(A)}{P(B)} $$
A probabilidade à priori, representada como P(A), é a probabilidade inicial da hipótese antes de observarmos qualquer evidência. Em termos de classificação, seria a probabilidade de um dado pertencer a uma determinada classe antes mesmo de olharmos para suas características. Por exemplo, se 30% dos e-mails em um dataset são spam, então a probabilidade a priori de um novo e-mail ser spam é de 30%.
A verossimilhança, representada como P(B|A), é a probabilidade de observar a evidência E dado que a hipótese “A” é verdadeira. No contexto de classificação, seria a probabilidade de observar um determinado conjunto de valores de features dado que o dado pertence a uma determinada classe. Por exemplo, qual é a probabilidade de um e-mail conter a palavra “promoção” dado que ele é spam?
A probabilidade marginal, representada como P(B), é a probabilidade total de observar a evidência E independentemente da hipótese. Ela funciona como um fator de normalização para garantir que as probabilidades calculadas somem 1.
A probabilidade à posteriori, representada como P(A|B), é o que queremos calcular: a probabilidade da hipótese ser verdadeira dado que observamos a evidência. É o resultado final que o algoritmo utiliza para tomar a decisão de classificação.
Florestas aleatórias
A floresta aleatória é um dos algoritmos de aprendizado de máquina mais poderosos e amplamente utilizados na indústria.
Ele pertence à família dos algoritmos de ensemble, que são métodos que combinam múltiplos modelos para obter um resultado mais robusto e preciso do que qualquer modelo individual conseguiria alcançar sozinho. O princípio fundamental por trás dessa abordagem é que a combinação de muitos modelos “fracos” ou “mediocres” resulta em um modelo coletivamente muito mais forte e confiável.
Como o próprio nome sugere, o algoritmo constrói uma floresta composta por múltiplas árvores de decisão. Cada árvore na floresta é treinada de forma ligeiramente diferente das outras, o que garante diversidade entre elas. Quando chega o momento de fazer uma predição, todas as árvores da floresta “votam” e a decisão final é tomada pela maioria. Esse mecanismo de votação é o que confere ao algoritmo sua robustez excepcional.
Além disso, é um algoritmo excepcionalmente versátil e robusto. Ele apresenta altíssima acurácia em uma ampla variedade de problemas sem necessitar de muito ajuste de hiperparâmetros. Lida muito bem com dados faltantes, valores extremos e features irrelevantes. Funciona igualmente bem para classificação e regressão.
Por fim, o algoritmo constrói centenas de árvores e requer considerável memória e poder de processamento, especialmente para bases de dados grandes. Além do mais, enquanto uma única Árvore de Decisão pode ser facilmente visualizada e interpretada, entender o raciocínio por trás da decisão de uma floresta com 200 árvores é praticamente impossível, tornando o modelo uma caixa-preta em termos de interpretabilidade.
Árvores extras
Árvores extras, cujo nome completo é Árvores Extremamente Aleatórias, é um algoritmo de ensemble muito próximo do florestas aleatórias, mas que leva o conceito de aleatoriedade a um nível ainda mais extremo. Foi proposto como uma alternativa que busca reduzir ainda mais a variância dos modelos através de uma aleatoriedade mais radical no processo de construção das árvores.
À primeira vista, o algoritmo árvores extras parece ser apenas uma versão mais aleatória do floresta aleatória, mas as implicações dessa aleatoriedade adicional são profundas e impactam diretamente nas características do modelo resultante, incluindo sua velocidade de treinamento e sua capacidade preditiva.
Comparação com a floresta aleatória
Para entender o que torna o algoritmo árvores extras verdadeiramente extremamente aleatório, é necessário comparar em detalhe como ele e o floresta aleatória tomam decisões de divisão nos nós.
No algoritmo floresta aleatória, quando é necessário decidir como dividir um nó, ele seleciona aleatoriamente um subconjunto de características e, para cada uma, busca o ponto de corte ótimo. Ou seja, ele testa vários valores possíveis e escolhe aquele que resulta na maior redução de impureza. Esse processo de busca pelo ponto de corte ótimo é computacionalmente custoso, pois exige avaliar múltiplos pontos de corte para cada característica considerada.
Já no algoritmo árvores extras, o processo é diferente em um aspecto crucial: para cada característica selecionada, ao invés de buscar o melhor ponto de corte, o algoritmo gera um ponto de corte completamente aleatório dentro do intervalo de valores daquela característica no conjunto de dados atual. Assim, a decisão de divisão é tomada com base no melhor corte entre esses melhores cortes aleatórios, e não entre os cortes ótimos. Essa mudança aparentemente simples tem consequências muito importantes para o comportamento do modelo.
Implementando em Python
Entendido todos os modelos, chegou a hora de implementa-lós. Nesta seção iremos organizar os dados de treino e teste e montar o código responsável por fazer a predição usando o Python.
Preparando o dataset
O primeiro passo de qualquer é a coleta e compreensão dos dados. Para este experimento, nós extraímos os dados reais de 50 proteínas hemaglutinina (HA) e 50 proteínas neuraminidase (NA), focando especificamente na variante H3N2.
Esses registros foram obtidos diretamente do banco de dados público do Instituto Nacional de Saúde dos Estados Unidos (NIH). O download é feito no padrão FASTA, um formato de texto amplamente utilizado na bioinformática.
Se você abrir um desses arquivos, notará que o material genético é representado por uma longa sequência de letras, onde cada caractere corresponde a uma das quatro bases nitrogenadas que compõem o DNA do vírus: Guanina (G), Citosina (C), Timina (T) e Adenina (A). A estrutura bruta do arquivo se parece exatamente com isso:
OM730361.1 |Influenza A virus (A/Florida/11/2021(H3N2)) segment 4 hemagglutinin (HA) gene, complete cds
GGATAATTCTATTAACCATGAAGACTATCATTGCTTTGAGCAACATTCTATGTCTTGTTTTCGCTCAAAAAATACCTGGAAATGACAATAGCACGGCAACGCTGTGCCTTGGGCACCATGCAGTACCAAACGGAACGATAGTGAAAACAATCACAAATGACCGAATTGAAGTTACTAATGCTACTGAGTTGGTTCAGAATTCATCAATAGGTGAAATATGCGGCAGTCCTCATCAGATCCTTGATGGAGGGAACTGCACACTAATAGATGCTCTATTGGGGGACCCTCAGTGTGACGGCT
Aqui encontramos o nosso primeiro obstáculo técnico de implementação. Os algoritmos da biblioteca Scikit-Learn são modelos matemáticos rigorosos. Eles não conseguem ler letras, strings ou interpretar texto livre; eles exigem matrizes numéricas para realizar seus cálculos.
Para transpor essa barreira, precisamos aplicar uma etapa de pré-processamento. Vamos implementar um conversor no nosso código Python que fará a varredura do arquivo FASTA e traduzirá cada caractere genético para um valor númerico correspondente.
O fluxo dessa conversão de texto biológico para dados tabulares numéricos funcionará conforme ilustrado abaixo:
Implementando o conversor
Entendido como o conversor funciona, podemos então implementá-lo usando o python. Criamos uma classe chamada EncodingData que faz a conversão exatamente como mostrado no trecho de código abaixo.
class EncodingData:
def encoding(self, sequence):
data_encoded = []
new_sequence = []
for i in sequence:
if i == 'A' :
data_encoded.append('1')
elif i =='T' :
data_encoded.append('2')
elif i =='G':
data_encoded.append('3')
elif i == 'C':
data_encoded.append('4')
for i in range(len(data_encoded)):
if i%15==0:
aux = data_encoded[i:i+15]
aux = ''.join(aux)
aux = float(aux)
new_sequence.append(aux)
return (new_sequence)
Separando dados de treino e teste
Como dito anteriormente, separamos 50 proteínas de HA e NA e com os dados genéticos devidamente convertidos para um formato numérico compreensível, a próxima etapa é preparar o terreno para ensinar os nossos modelos. É importante destacar que, focamos nas amostras das proteínas HA e NA da cepa H3N2, coletadas especificamente entre os anos de 2021 e 2022.
Para garantir que os algoritmos realmente aprendam os padrões biológicos das mutações e não apenas “decorem” as informações, aplicamos a divisão clássica do aprendizado de máquina: 70% dos dados foi destinado para a fase de treinamento e os 30% restantes foram isolados exclusivamente para os testes de validação.
Após o agrupamento e a separação, nossa base estruturada resultou em quatro arquivos finais prontos para serem processados pelo Python: dois arquivos referentes à hemaglutinina (treino e teste) e dois referentes à neuraminidase (treino e teste). A tabela abaixo apresenta uma amostra das cepas utilizadas no experimento, detalhando informações cruciais para a nossa análise, como o local e o ano exato de cada coleta.
| Local | Ano | Segmento | Dados |
|---|---|---|---|
| Flórida | 2021 | H3N2-HA | Treinamento |
| Califórnia | 2022 | H3N2-HA | Teste |
| Pensilvânia | 2021 | H3N2-NA | Treinamento |
| Califórnia | 2022 | H3N2-NA | Teste |
Desenvolvimento dos modelos com Scikit-Learn
A partir de agora, iremos implementar as classes de todos os modelos que discutimos até aqui.
Árvores de decisão
from sklearn import tree
from sklearn import metrics
from sklearn.model_selection import cross_val_score, train_test_split
class DecisionTree:
def __init__(self, X, Y):
decisionTree = tree.DecisionTreeRegressor()
decisionTree.fit(X, Y)
score = cross_val_score(decisionTree, X, Y, cv=2)
print('Decision Tree score: ', score)
print('Average Accuracy: %0.2f (+/- %0.2f)' % (score.mean()*100, score.std()*100))
X_train, X_test, Y_train, Y_test = train_test_split(X,Y,test_size=0.3,random_state=50)
decisionTree.fit(X_train,Y_train)
print('Score ', decisionTree.score(X_test,Y_test))
y_pred_dtr = decisionTree.predict(X_test)
print('----- Metrics to Decision Trees-----')
print('R2 score:', metrics.r2_score(Y_test,y_pred_dtr,multioutput='variance_weighted'))
print('Mean Absolute Error:', metrics.mean_absolute_error(Y_test, y_pred_dtr))
print('Mean Squared Error:', metrics.mean_squared_error(Y_test,y_pred_dtr))
Naive Bayes
from sklearn import metrics
from sklearn.model_selection import cross_val_score, train_test_split
from sklearn.naive_bayes import GaussianNB
import numpy as np
class NaiveBayes:
def __init__(self, X, Y):
naiveBayes = GaussianNB()
X = np.array(X)
Y = np.array(Y)
X = X.reshape(-1, 1)
Y = Y.reshape(-1, 1)
naiveBayes.fit(X, np.ravel(Y, order='C'))
score = cross_val_score(naiveBayes, X, np.ravel(Y, order='C'), cv=2)
print('Naive Bayes', score)
print("Average Accuracy: %0.2f (+/- %0.2f)" % (score.mean()*100, score.std() *100))
X_train,X_test,y_train,Y_test = train_test_split(X,Y,test_size=0.3,random_state=50)
naiveBayes.fit(X_train,np.ravel(y_train, order='C'))
print('Score', naiveBayes.score(X_test,Y_test))
y_pred_nb = naiveBayes.predict(X_test)
print('----- Metrics to Naive Bayes-------')
print('R2 score:', metrics.r2_score(Y_test,y_pred_nb,multioutput='variance_weighted'))
print('Mean Absolute Error:', metrics.mean_absolute_error(Y_test, y_pred_nb))
print('Mean Squared Error:', metrics.mean_squared_error(Y_test,y_pred_nb))
Florestas aleatórias
from sklearn import ensemble, metrics
from sklearn.model_selection import cross_val_score, train_test_split
class RandomForest:
def __init__(self, X, Y):
randomForest = ensemble.RandomForestRegressor(n_estimators=23)
randomForest.fit(X, Y)
score = cross_val_score(randomForest, X, Y, cv=2)
print('Random Forest', score)
print("Average Accuracy: %0.2f (+/- %0.2f)" % (score.mean()*100, score.std() *100))
X_train, X_test, Y_train, Y_test = train_test_split(X,Y,test_size=0.3,random_state=50)
randomForest.fit(X_train,Y_train)
print('Score ', randomForest.score(X_test,Y_test))
y_pred_rfr = randomForest.predict(X_test)
print('----- Metrics to Random Forest-----')
print('R2 score:', metrics.r2_score(Y_test,y_pred_rfr,multioutput='variance_weighted'))
print('Mean Absolute Error:', metrics.mean_absolute_error(Y_test, y_pred_rfr))
print('Mean Squared Error:', metrics.mean_squared_error(Y_test,y_pred_rfr))
Árvores extras
from sklearn import metrics
from sklearn.model_selection import cross_val_score, train_test_split
from sklearn import ensemble
class ExtraTrees:
def __init__(self, X, Y):
extraTrees = ensemble.ExtraTreesRegressor(n_estimators=3)
extraTrees.fit(X, Y)
score = cross_val_score(extraTrees, X, Y, cv=2)
print('Extra Trees', score)
print("Average Accuracy: %0.2f (+/- %0.2f)" % (score.mean()*100, score.std() *100))
X_train,X_test,y_train,Y_test = train_test_split(X,Y,test_size=0.3,random_state=50)
extraTrees.fit(X_train,y_train)
print('Score', extraTrees.score(X_test,Y_test))
y_pred_ext = extraTrees.predict(X_test)
print('----- Metrics to Extra Trees-----')
print('R2 score:', metrics.r2_score(Y_test,y_pred_ext,multioutput='variance_weighted'))
print('Mean Absolute Error:', metrics.mean_absolute_error(Y_test, y_pred_ext))
print('Mean Squared Error:', metrics.mean_squared_error(Y_test,y_pred_ext))
Treinando os modelos com Scikit-Learn
Com as classes de todos os algoritmos criadas e o nosso conversor pronto, chegou a hora de unirmos todas as peças no nosso arquivo principal de execução.
Neste script, utilizaremos a poderosa biblioteca Biopython para ler os nossos arquivos FASTA de maneira limpa e eficiente. O código fará a extração das sequências, passará os dados brutos pelo nosso EncodingData para transformá-los em matrizes numéricas e, finalmente, instanciará as classes dos quatro modelos para iniciar a “batalha”.
Para facilitar os testes, deixamos o código modular. No exemplo abaixo, o script está configurado para ler os dados da proteína neuraminidase. Caso queira testar a Hemaglutinina, basta comentar as linhas referentes à NA e descomentar as linhas da HA.
import encoding_data
from Bio import SeqIO
from prediction_model.decisionTree import DecisionTree
from prediction_model.extraTrees import ExtraTrees
from prediction_model.naiveBayes import NaiveBayes
from prediction_model.randomForest import RandomForest
def main():
# Para testar a proteína HA, descomente as linhas abaixo e comente as da NA
# file_train = open('flu-data/H3N2/HA/H3N2-HA50-2021.fasta')
# file_test = open('flu-data/H3N2/HA/H3N2-HA50-2022.fasta')
# Configuração atual: Proteína NA
file_train = open('flu-data/H3N2/NA/H3N2-NA50-2021.fasta')
file_test = open('flu-data/H3N2/NA/H3N2-NA50-2022.fasta')
data_train = list(SeqIO.parse(file_train, 'fasta'))
data_test = list(SeqIO.parse(file_test, 'fasta'))
train = []
test = []
X = []
Y = []
for i in range(len(data_train)):
train.append(data_train[i].seq)
for j in range(len(data_test)):
test.append(data_test[j].seq)
encode = encoding_data.EncodingData()
for k in range(len(train)):
encoded_train = encode.encoding(train[k])
X.append(encoded_train)
for l in range(len(test)):
encoded_test = encode.encoding(test[l])
Y.append(encoded_test)
print('--- Avaliando Modelos ---')
DecisionTree(X, Y)
print('\n')
RandomForest(X, Y)
print('\n')
ExtraTrees(X, Y)
print('\n')
NaiveBayes(X, Y)
if __name__ == '__main__':
main()
Analisando os resultados
Agora, com tudo implementado, podemos executar nosso algoritmo usando o comando
python3 main.py
Quando executar esse comando, alguns problemas podem acontecer, esses problemas são listados abaixo.
- ModuleNotFoundError: No module named ‘Bio’
- ModuleNotFoundError: No module named ‘sklearn’
Isso acontece porque a aplicação usa bibliotecas externas e elas precisam ser baixadas para que funcione corretamente. Para corrigir aplique a correção abaixo
- pip3 install Bio
- pip3 install scikit-learn
Feito isso, rode novamente e aplicação irá executar.
Comparativo de acurácia
Agora, analisaremos o resultado de cada algoritmo para HA e NA.
Desempenho da predição de HA
Quando executamos o algoritmo para a proteína HA obtemos o resultado apresentado abaixo.
Desempenho da predição de NA
Já quando executamos o algoritmo para a proteína NA obtemos o resultado apresentado abaixo.
O algoritmo floresta aleatória foi a estrela do experimento. Na proteína Hemaglutinina (HA), ele alcançou uma excelente acurácia de 91.41% e o melhor R² de 0.896. Na Neuraminidase (NA), o padrão se repetiu com um R² de 0.879. Mais importante ainda: os erros absolutos (MAE) e quadráticos (MSE) foram, de longe, os menores entre todos os concorrentes, provando que suas predições são consistentes e erraram por muito pouco.
Já o algoritmo árvore de decisão e as árvores extras tiveram um desempenho razoável, mas ficaram presas na casa dos 0.77 de R² para HA e 0.68 para NA. Nas árvores extras testando a NA, notamos um detalhe interessante: a acurácia média de validação cruzada foi alta (93.82%), mas o score final no teste caiu para 0.689, um indício clássico de que o modelo decorou os dados de treino, mas teve mais dificuldade com dados novos.
Por fim, o Naive Bayes nos deu uma excelente lição prática de Ciência de Dados. Ao olharmos para as métricas de erro, os números do Naive Bayes explodiram para escalas astronômicas. Por que isso aconteceu? O Naive Bayes assume que todas as características são 100% independentes umas das outras. Na biologia, isso não é verdade: a sequência de uma proteína segue uma ordem altamente dependente. Além disso, o modelo espera distribuições normais, e nós entregamos categorias numéricas. O algoritmo simplesmente não conseguiu lidar com a estrutura biológica dos dados.
Qual é a melhor escolha ?
Diante de todos os testes, validações e métricas de erro, o veredito é inquestionável: o algoritmo floresta aleatória é a melhor escolha para prever as mutações do vírus Influenza.
A complexidade da biologia molecular exige um algoritmo que consiga enxergar nuances sem se viciar nos dados de treino. Como o algoritmo vencedor cria múltiplas árvores diferentes e toma sua decisão baseada em um consenso, ele conseguiu neutralizar o “ruído” genético e encontrar os verdadeiros padrões de mutação das proteínas HA e NA com quase 90% de confiança preditiva.
Nenhum conteúdo publicado neste site constitui, em circunstância alguma, aconselhamento financeiro, recomendação de investimento, compra e venda de ativos ou qualquer tipo de orientação médica. Para decisões financeiras ou de saúde, o utilizador deve sempre consultar profissionais devidamente certificados e credenciados nas respetivas áreas.


